1.2 Drylab Resource Overview
Get Started

There are countless resources built into Drylab to make your analysis sharper, faster, and more scientifically accurate. From databases and accelerated tools to full pipelines, pre-installed packages, and skill-based workflows — each layer helps the AI understand your intent and deliver results that align with real-world best practices.
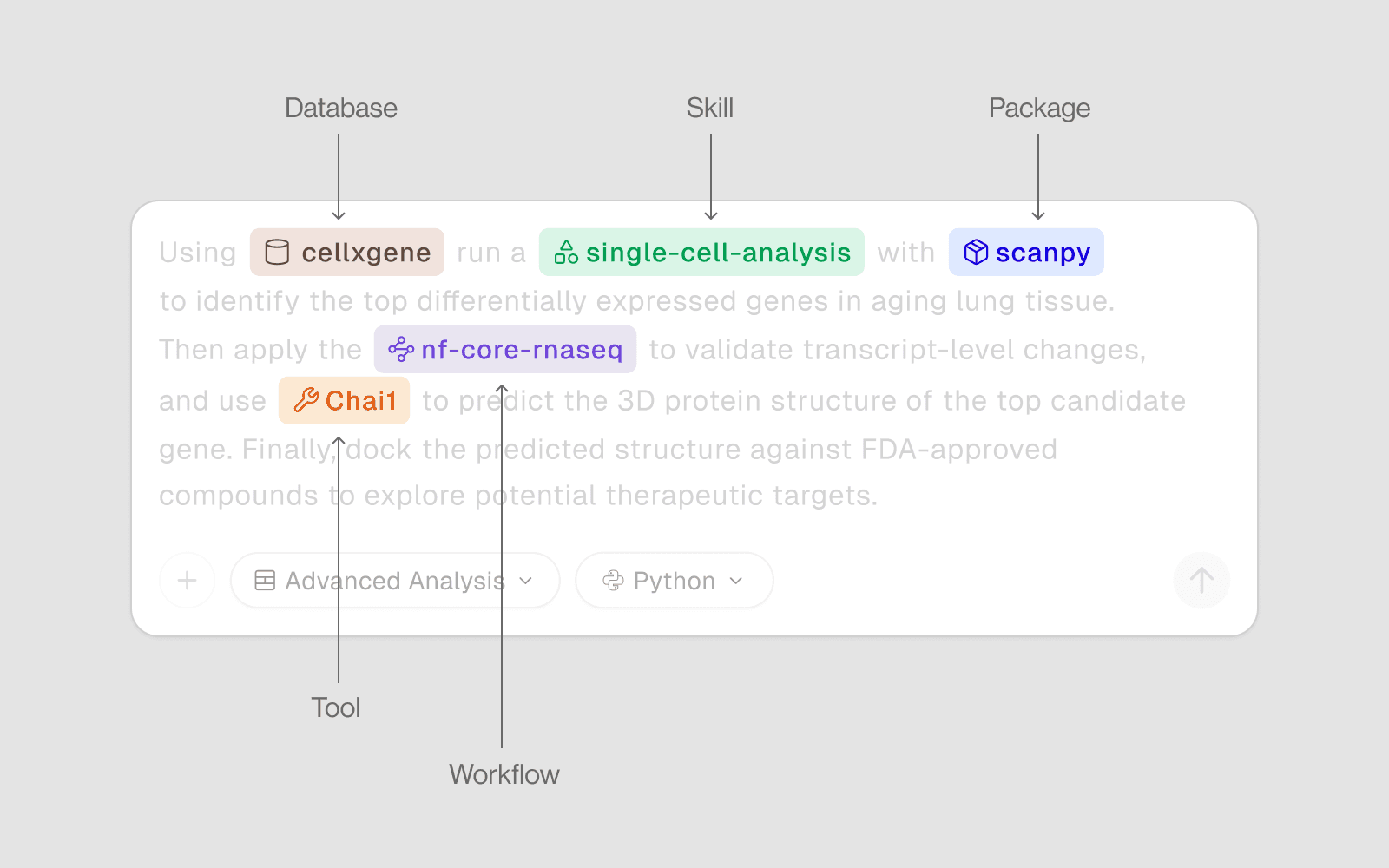
Databases | Query biological data (no compute needed ) |
Accelerated Tools | Run GPU/HPC jobs (protein folding, docking) |
Pipelines | End-to-end Nextflow workflows (RNA-seq, variant calling) |
Pipelines | Python/R libraries (pre-installed, ready to use) |
Skills | Step-by-step analysis guides (best practice workflows) |
Overview
By using @ to call these resources directly in chat, you can guide the AI to use the right method or dataset — making your output more precise, reproducible, and perfectly tuned to your expectations.
1. Databases
What they are: Connections to 80+ curated biological databases. Query them in plain English — no API keys, no manual HTTP requests.
When to use: When you need reference data — gene info, protein structures, drug targets, variants, pathways, expression data.
Category | Key Databases |
|---|---|
Protein | UniProt, PDB, AlphaFold |
Genomics | Ensembl, gnomAD, ClinVar |
Pathways | KEGG, Reactome |
Drugs | DrugBank, PubChem, ChEMBL |
Cancer | cBioPortal, OncoKB |
Expression | GTEx, GEO |
Single-cell | CellxGene |
2. Accelerated Tools
What they are:
50+ GPU/HPC-powered compute tools for heavy scientific tasks — protein folding, molecular docking, genome alignment, and variant calling.
When to use:
When computation is too intensive for a standard CPU notebook — structure prediction, large-scale docking, or long-read assembly.
Category | Tools |
|---|---|
Protein Structure Prediction | Boltz2, Chai-1, ESMFold, OmegaFold, OpenFold3, RosettaFold3, IntelliFold |
Antibody & Immunology | ABodyBuilder 3, AbLang, AbLang-MPNN, AbMPNN, AbGPT, AbMAP, ADAPT -- Antigen-receptor Design Against Peptide-MHC Targets, AntiFold, AntiBERTy, ANARCI, DeepRank-Ab, IgDesign, ImmuneBuilder, RFantibody, TCRmodel2 |
Protein Design & Engineering | BindCraft, Protein MPNN, RFdiffusion3, RFpeptides, PXDesign, Profluent E1, EvoEF2, mBER (Manifold Binder Engineering and Refinement), Protein Gibbs Sampler (pgen), Protein Scoring (COMPSS / pgen), ProteinX, ProteusAl, SaProt |
Molecular Docking & Drug Discovery | DiffDock, EquiBind, FlowDock, SurfDock, TankBind, QVina (QuickVina 2 / QuickVina-W), rDock, Uni-Mol Docking V2, Pocket2Mol, Roshambo (shape-based screening), AF2BIND (Binding Site Prediction), DockQ |
Molecular Dynamics & Simulation | GROMACS, OpenMM, LAMMPS, gmx_MMPBSA (Free Energy Calculations), GPUMD, Tinker, Tinker-GPU (Tinker9), HOOMD-blue, UAMMD, Molly. jl |
Protein/Molecular Representation & Analysis | Uni-Mol, Aggrescan3D (A3D), AllMetal3D (Metal lon Binding, Site Prediction), SpatialPPlv2, Protein Hunter |
RNA Structure & Design | RhoFold, RhoDesign, RiboDiffusion, RNAPrO |
Sequencing & Alignment | STAR, HISAT2, Bowtie2, Minimap2, Kallisto, Kallisto GPU, Salmon, StringTie, Dorado (Oxford Nanopore basecaller) |
Genome Assembly | Flye, Hifiasm, SPAdes, MEGAHIT, NextDenovo, Raven, Trinity, Unicycle, Verkko, WTDBG2 |
Variant Calling | BCFtools, Clair3, FreeBayes, Longshot, Medaka, NanoCaller, PEPPER-Margin-DeepVariant, Sniffles, Strelka2, VariantFormer |
Single-Cell & Transcriptomics AI | rapids_singlecell, scvi-tools, SCGPT, SPRINT, TranscriptFormer, UCE (Universal Cell Embeddings) |
Genomics & Regulatory | AlphaGenome, AlphaGenome |
QC & Reporting | MultiQC |
Sample: Predict the 3D structure of this sequence using Chai-1: MKTAYIAKQR...
3. Pipelines
What they are:
84+ production-ready Nextflow (nf-core) workflows for full upstream bioinformatics processing — from raw FASTQ to analysis-ready output.
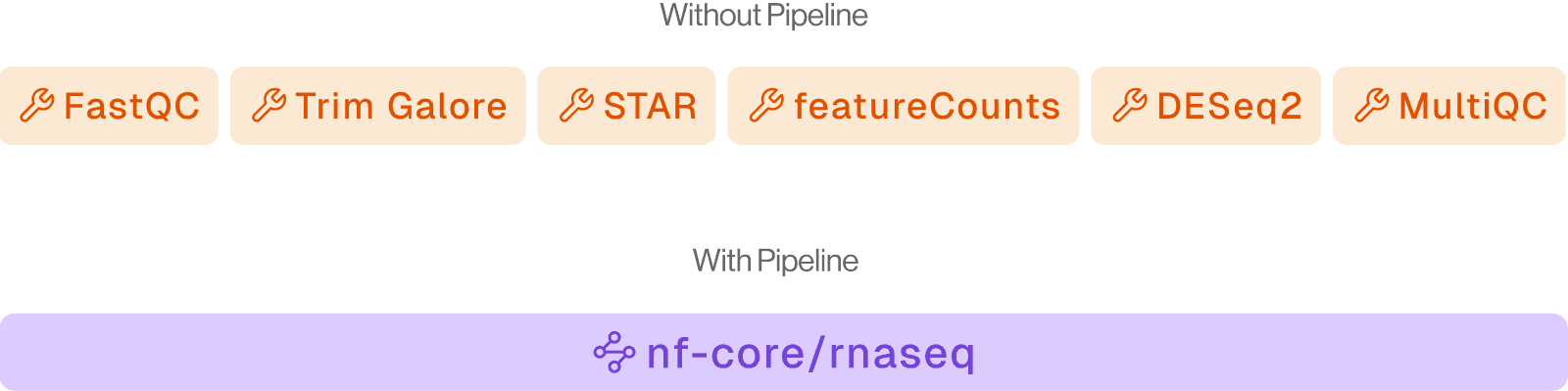
When to use:
When you have raw sequencing data and need standardized, reproducible processing at scale.
Category | Pipelines |
|---|---|
Bulk RNA-seq | nf-core-rnaseq |
Single-cell | nf-core-scrnaseq |
Variant Calling | nf-core-sarek |
Amplicon / 16S | nf-core-ampliseq |
ChIP-seq / ATAC-seq | nf-core-chipseq, nf-core-atacseq |
Metagenomics | nf-core-mag, nf-core-taxprofiler |
Sample: Run nf-core-rnaseq on my samplesheet at drylab://My Project/data/samplesheet.csv with genome GRCh38.
4. Packages
What they are:
Pre-installed Python and R libraries covering the full scientific computing stack — ready to import with no setup.
When to use:
For all interactive analysis in the notebook — data manipulation, statistics, machine learning, visualization.
R packages (via Rscript or R cells):
Seurat | Single Cell |
DESeq2 | Differential expression |
ggplot2 | Visualization |
edgeR | RNA-seq |
limma | Linear models |
Sample: Use Scanpy to run PCA and Leiden clustering on my h5ad file with resolution 0.5.
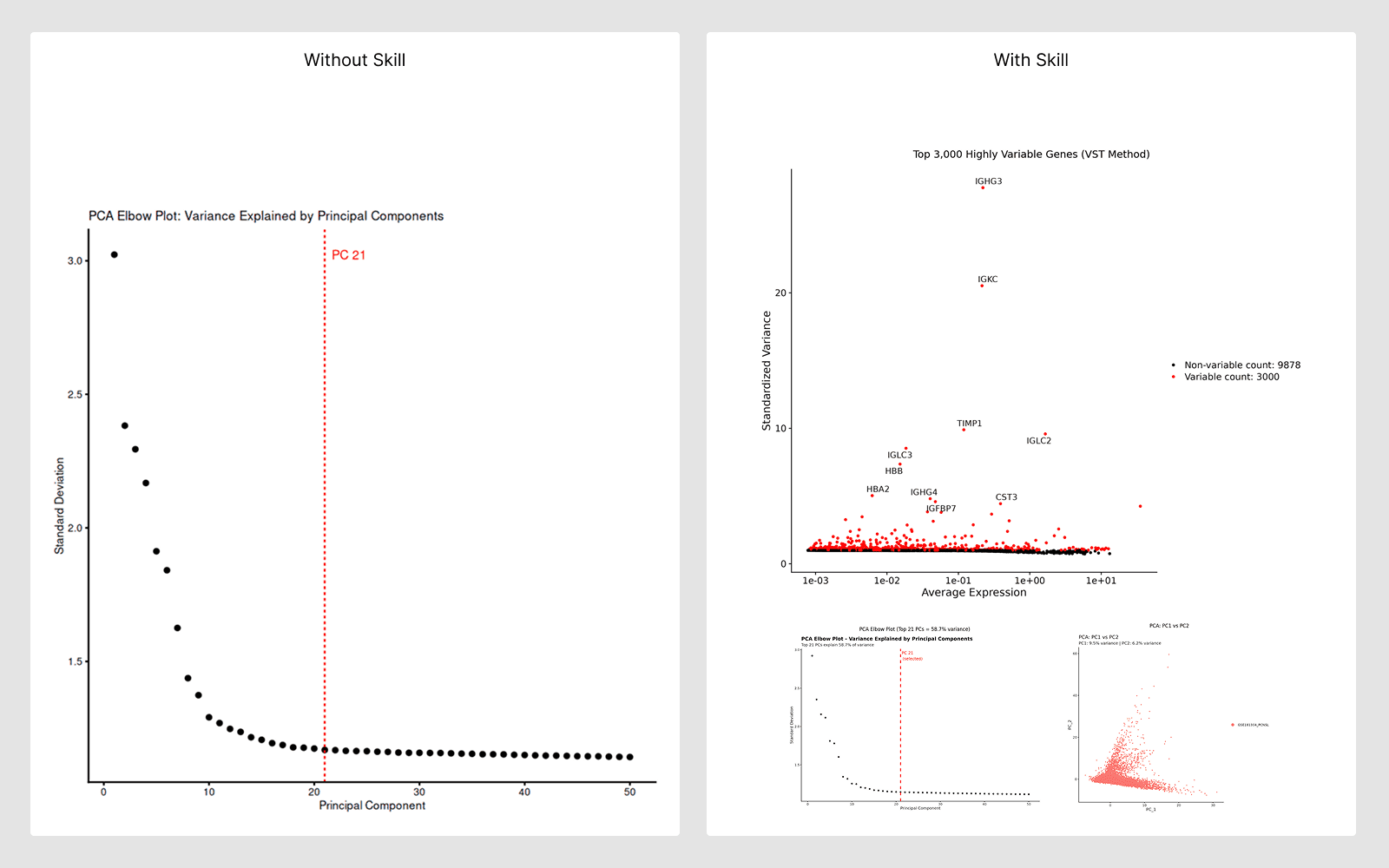
5. Skills
What they are:
Curated, step-by-step analysis guides encoding best practices for specific workflows. The AI reads these guides automatically and follows validated protocols.
When to use:
When doing a well-established analysis type — the AI follows the skill guide to ensure scientifically correct, reproducible results.
Domain | Example Skills |
|---|---|
Single-cell | QC, clustering, annotation, DE, trajectory |
Spatial | Spatially variable genes, deconvolution |
Bulk RNA-seq | Normalization, DE, pathway enrichment |
Proteomics | Structure prediction, docking |
Phylogenomics | Tree reconstruction, alignment |
How the AI uses skills:
Automatically discovers the relevant skill for your task
Reads the protocol and selects the right methods and parameters
Validates results against the skill’s quality checkpoints
Sample: Run single-cell QC and clustering following best practices on my h5ad file.
How to Use All Resources Effectively
Let the AI discover resources for you
“I have scRNA-seq data. What tools and databases should I use for cell type annotation and differential expression?”
Chain resources together
Query CellxGene for a human lung reference atlas → Database
Align FASTQ files with STAR → Accelerated Tool
Run nf-core-scrnaseq → Pipeline
Cluster and annotate using Scanpy + best practices → Package + Skill
Query DrugBank for drugs targeting identified markers → Database
