
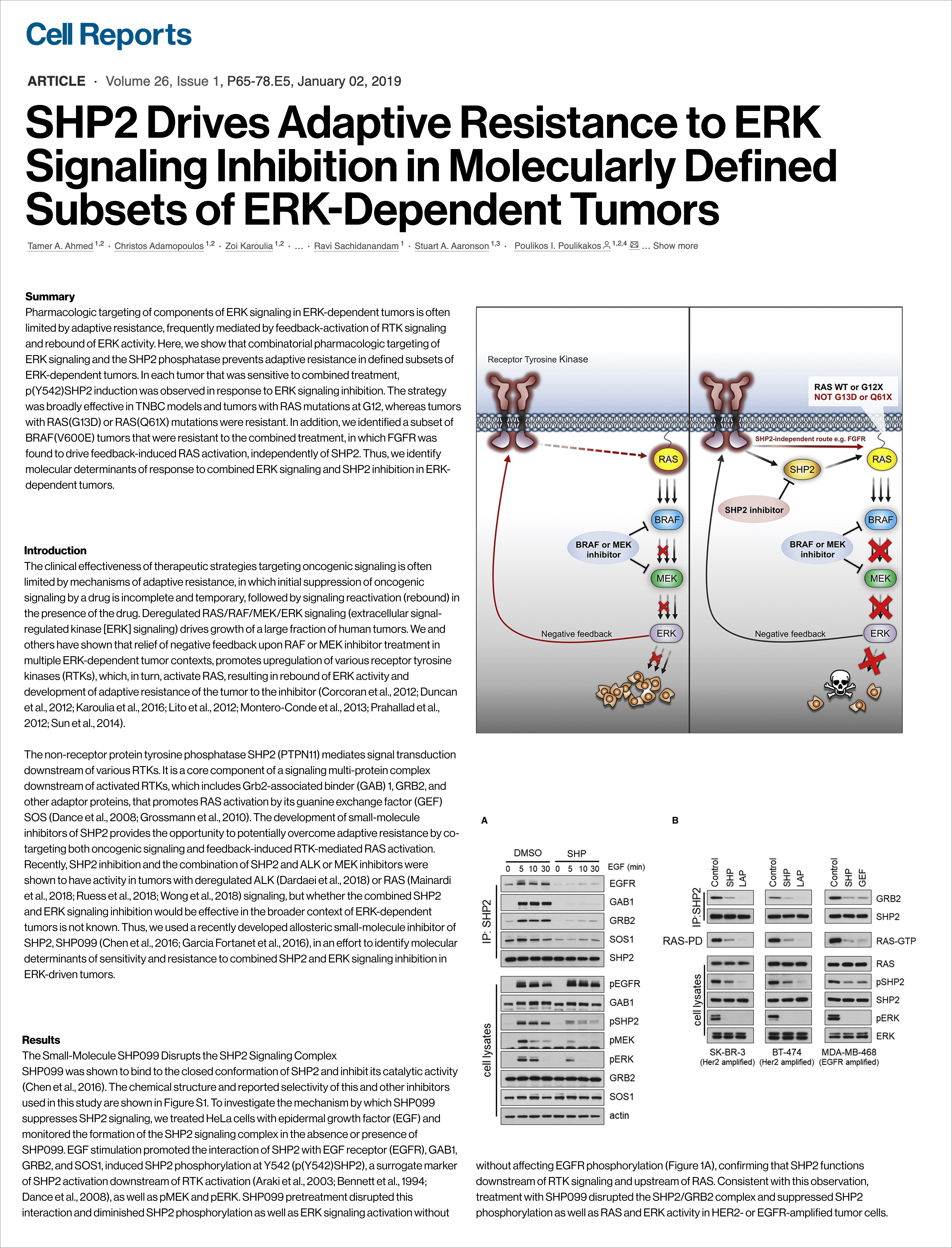
原始论文假设
SHP2通过将RTK驱动的负反馈与RAS再激活耦合,介导RAF/MEK抑制剂的适应性抵抗。结合SHP2 + ERK抑制可以克服这一点,并且p(Y542)SHP2 + RAS突变亚型可以预测反应。
数据集
使用来自GEO (GSE121117) 的RNA-seq数据作为主要和唯一的数据集。
原始RNA-seq实验中仅有3个细胞系:HTH104, sw1736, WIDR
干实验室分析
间接证实了论文中描述的分子亚型区分:HTH104/sw1736 (SHP2阴性,FGFR依赖型) 与WIDR (SHP2阳性,EGFR依赖型) 的分裂在转录数据中完全再现——这两个组在PC1上分离,层次聚类和RTK基因热图中。
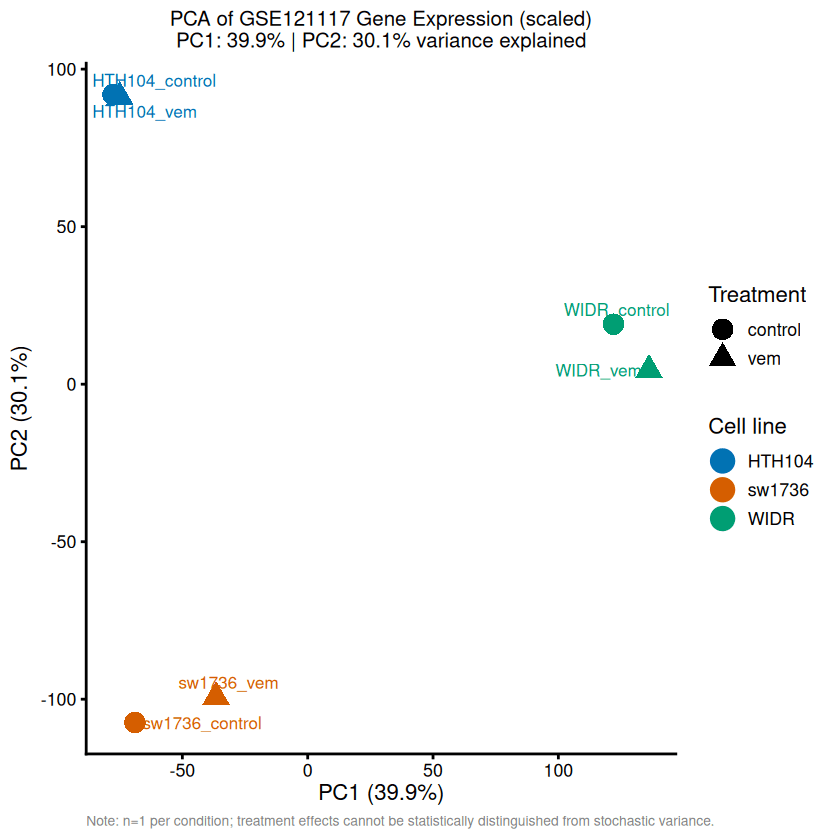
PCA和层次聚类
PC1 (39.9%) 和PC2 (30.1%) 共解释了总方差的70.0%。经过缩放后,方差比未缩放分析分布更均匀(PC1+PC2为85.9%),表明之前几个高方差基因膨胀了PC1。
PC1主要通过原始组织分离样本:WIDR (结直肠) 与HTH104和SW1736 (甲状腺ATC) 聚集分开。值得注意的是,尽管SW1736和WIDR都携带BRAF-V600E突变,但它们处于相反的分支,建议突变状态不是转录组变异的主要驱动因素。相反,细胞系身份占据主导,而处理效果相对较为微妙。
在每个细胞系中,控制和vemurafenib样品紧密聚集,表明细胞系之间的基线转录组差异超过了药物诱导的变化。
这种模式与适应性抵抗主要通过翻译后信号再激活发生一致。
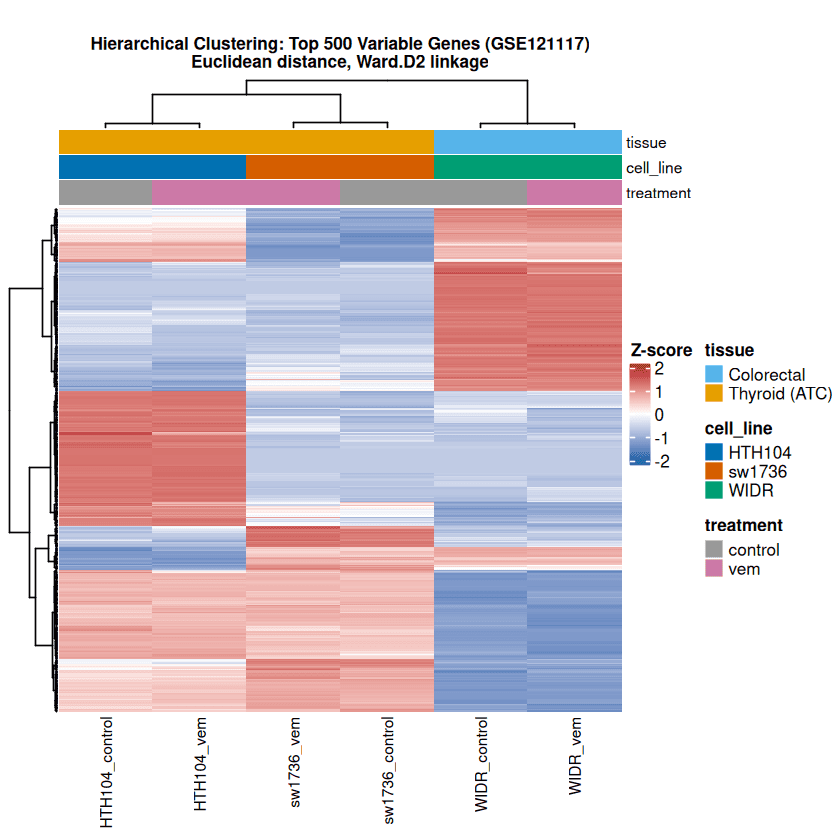
柱状树状图显示甲状腺(ATC)与结直肠样本之间有一个明显的主要分裂。
出现了两个主要基因程序:一个在HTH104和SW1736中上调但在WIDR中抑制的块,另一个在WIDR中强烈表达。这个二分模式与组织起源和SHP2依赖性一致,而不是突变背景,因为所有三个系都携带BRAF-V600E。
vemurafenib治疗在每个细胞系中仅引起微妙的变化,与PCA结果和n = 1设计一致,后者阻止了处理效果的正式统计测试。
RTK信号基因分析
从文献中汇编了一组159个RTK信号基因(受体、配体、RAS调节因子、细胞因子、趋化因子),
MSigDB Reactome/KEGG路径和GSEA领先基因。基因映射到数据集。计算行式Z分数跨六个样本,并限制在±2.5 SD以内。组织差异分数(WIDR平均Z − 甲状腺平均Z)量化了谱系偏差。使用ComplexHeatmap和Ward.D2层次聚类生成热图。
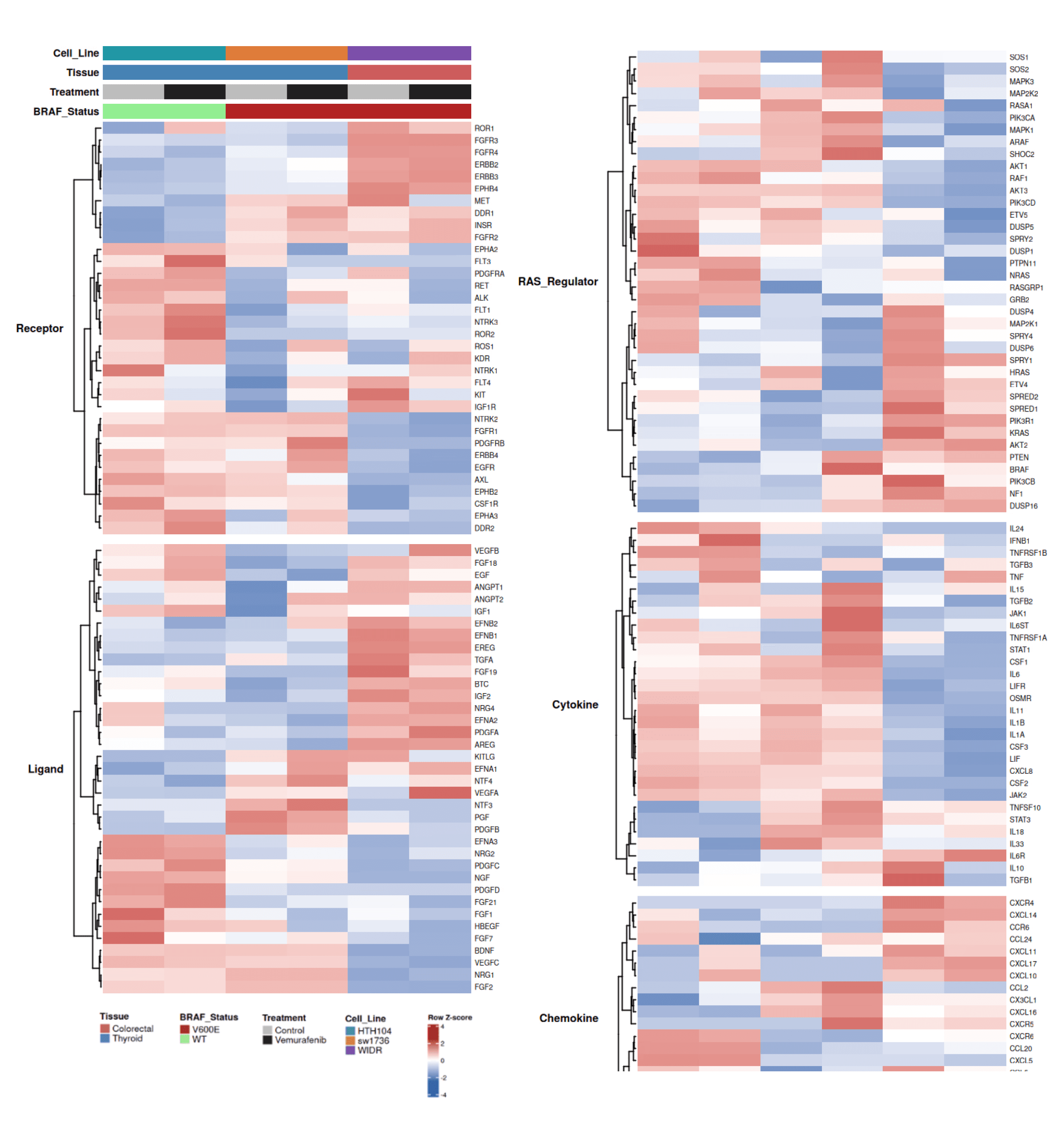
•基因覆盖率:受体、配体和RAS调节因子的覆盖率高(97-100%);细胞因子/趋化因子较低(53-79%),与上皮细胞系中的有限免疫基因表达一致。
•组织特异性RTK程序:一个明显的二分模式分离甲状腺ATC和结直肠WIDR。WIDR富集基因包括EREG, FGFR3, CXCR4, EFNB1, EPHB4; ATC富集基因包括BDNF, OSMR, FGFR1, AKT3, VEGFC。
•受体差异:FGFR3/4和ERBB2/3在WIDR中更高,而FGFR1和IGF1R在ATC中更高。
•配体差异:EGFR配体(AREG, EREG, TGFA)在WIDR中富集,而FGF2, BDNF, VEGFC在ATC中富集。
•RAS反馈调节因子:观察到明显的MAPK反馈布线(例如SPRY1在WIDR中富集)。
RTK信号程序因谱系而异。ATC系表现出FGFR1-FGF2轴,而WIDR表现出FGFR3/4和EGFR配体程序(AREG/EREG/ERBB3),与EGFR-SHP2信号一致。额外的PI3K/AKT同工型差异(ATC中的PIK3CD/AKT3对比WIDR中的PIK3R1)建议平行生存途径。
FGFR/ErbB家族和SHP2/GRB2/RAS分析
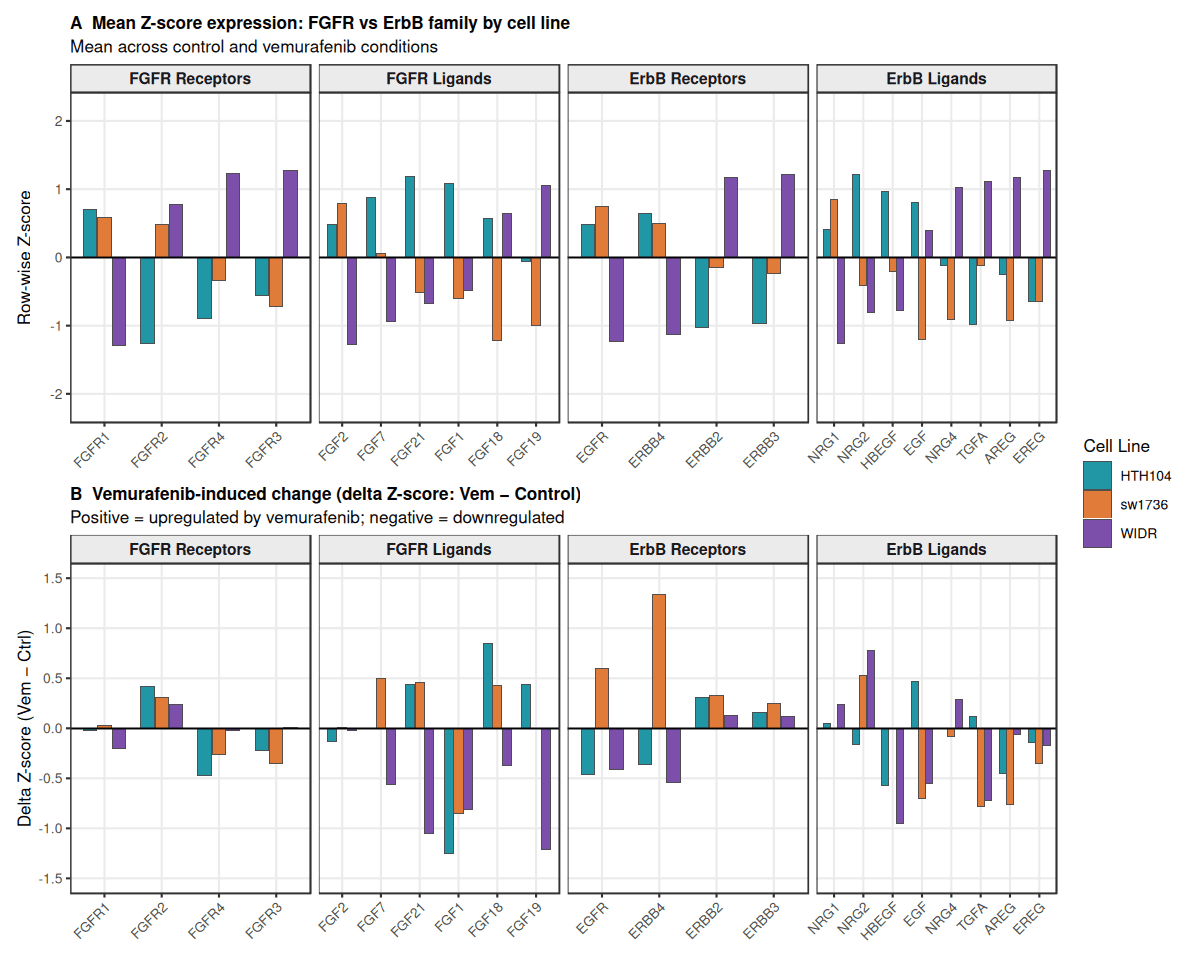
子图A – 基线RTK程序
基线表达揭示出明显的谱系特异的RTK信号差异。甲状腺系(HTH104, SW1736)显示出更高的FGFR信号,特别是FGFR1及其配体FGF2,与FGFR驱动程序一致。
相反,WIDR表现出更强的ErbB网络活动,包括ERBB3和EGFR配体(AREG, EREG),伴随着更高的FGFR3/4表达。此模式反映了FGFR1(甲状腺)→ FGFR3/4(结直肠)同工型转换,与已知肿瘤生物学一致。
子图B – Vemurafenib响应
ΔZ分数的分析显示出对BRAF抑制的不同适应性反应。甲状腺模型显示FGFR配体的转录变化,主要体现在补偿性的RTK信号发生在ERK抑制后。相反,WIDR显示最小的转录变化,暗示抵抗可能通过翻译后信号机制(例如,ERBB3-SHP2耦合)而不是大规模转录转变发生。
总体来看,结果显示ERK路径抑制触发了谱系特异性的RTK反馈程序:甲状腺模型优先激活FGFR信号,而结直肠模型更多依赖于ErbB网络,支持Ahmed等人, 2019中描述的肿瘤特异性适应性抵抗机制。
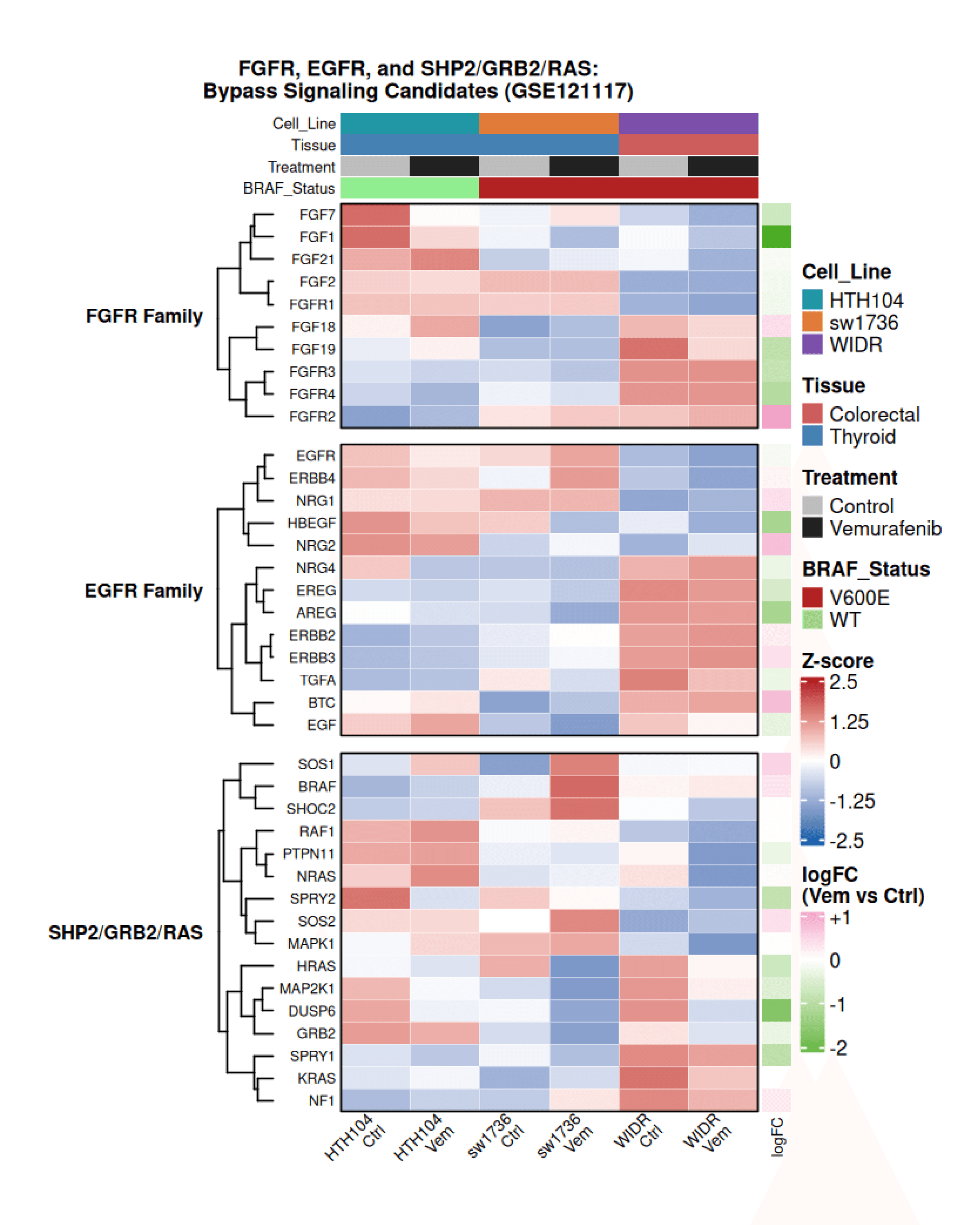
为探究ERK抑制后潜在的绕过信号,我们检查了涉及FGFR, EGFR/ErbB和SHP2/GRB2/RAS信号的基因。
行聚类的表达揭示出明显的谱系特异模式。甲状腺衍生系(HTH104, SW1736)显示出更高的基线FGFR配体和受体表达,而结直肠系WIDR则表现出更强的EGFR/ErbB路径活动。
在vemurafenib治疗后,甲状腺模型维持或增加FGFR信号,而WIDR则表现出较强的EGFR/ErbB成分和下游RAS信号激活。SHP2/GRB2/RAS基因的差异表达进一步支持一个模型,其中MAPK抑制触发肿瘤特异性的RTK驱动绕过信号,与Ahmed等人, 2019中描述的机制一致。
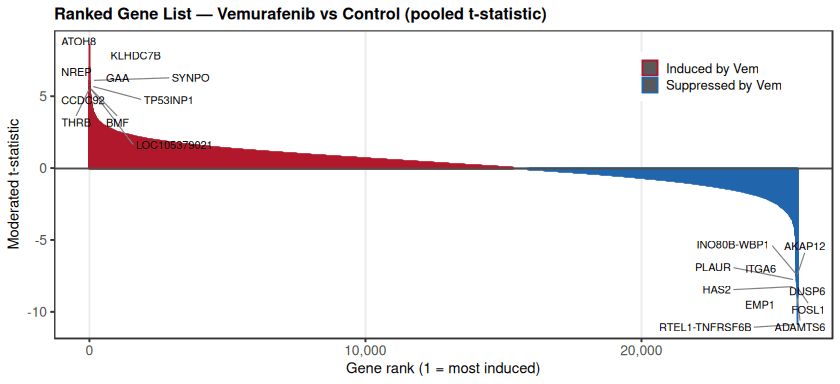
差异表达分析和基因集富集分析(GSEA)
在vemurafenib处理和控制样本之间的全基因组差异基因表达排名进一步揭示了ERK通路抑制引起的全球转录群体变化。
排名分布显示出明显的不对称性,一组基因被vemurafenib强烈诱导(正t统计量),而另一组基因被强烈抑制(负t统计量)。在最显著诱导的基因中有ATOH8, KLHDC7B, SYNPO, TP53INP1, 和NREP,暗示ERK通路抑制后压力反应和转录适应程序的激活。
相反,基因如FOSL1, DUSP6, HAS2, ITGA6, PLAUR, 和ADAMTS6是最强烈下调的,其中许多与ERK/MAPK信号输出或细胞外基质重塑有关。总体分布表明BRAF抑制未能产生统一的转录抑制,而是重新编程了一组特定的与信号反馈和细胞适应相关的基因。
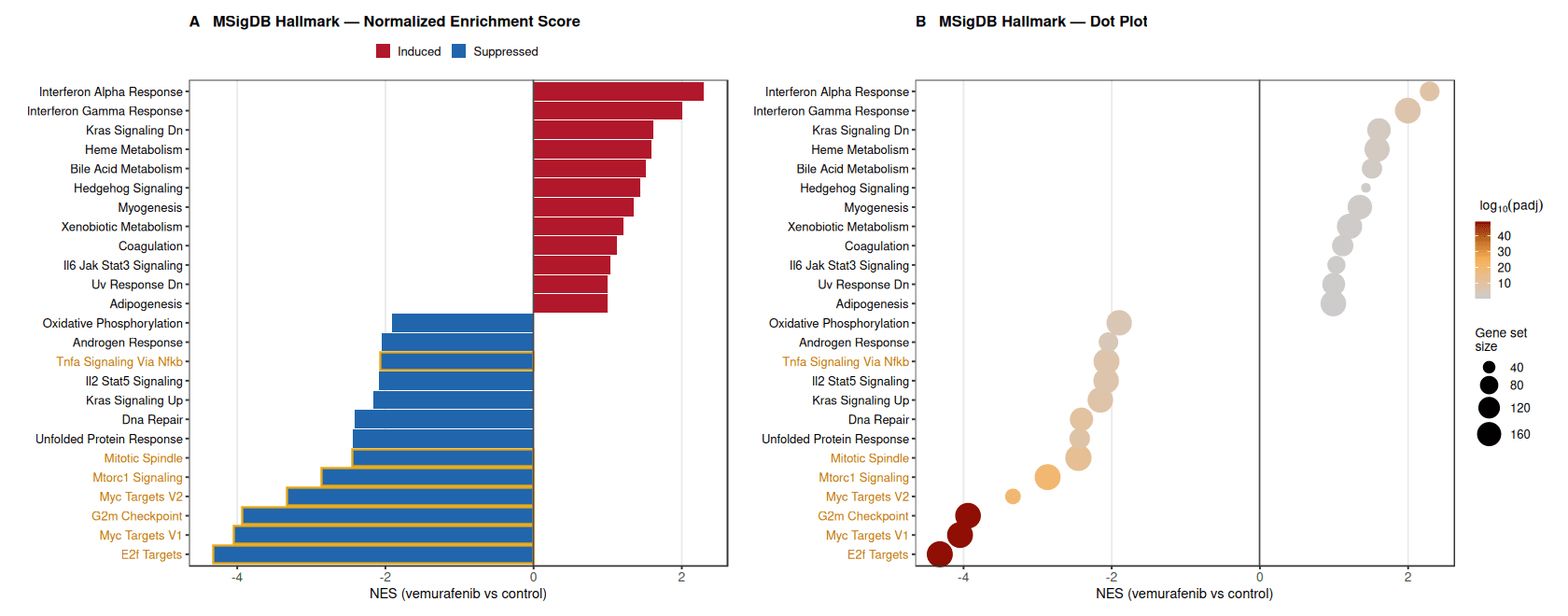
标志性通路分析揭示了在ERK路径抑制后显著被诱导和抑制的路径之间的明显分离。
免疫和应激相关路径,包括干扰素Alpha响应、干扰素Gamma响应和IL6-JAK-STAT3信号,在治疗后显著富集于上调基因中。
相反,细胞增殖和增长控制相关路径被强烈抑制,包括E2F Targets, MYC Targets (V1/V2), G2M Checkpoint和MTORC1信号,表明在BRAF抑制后增殖转录程序总体减少。
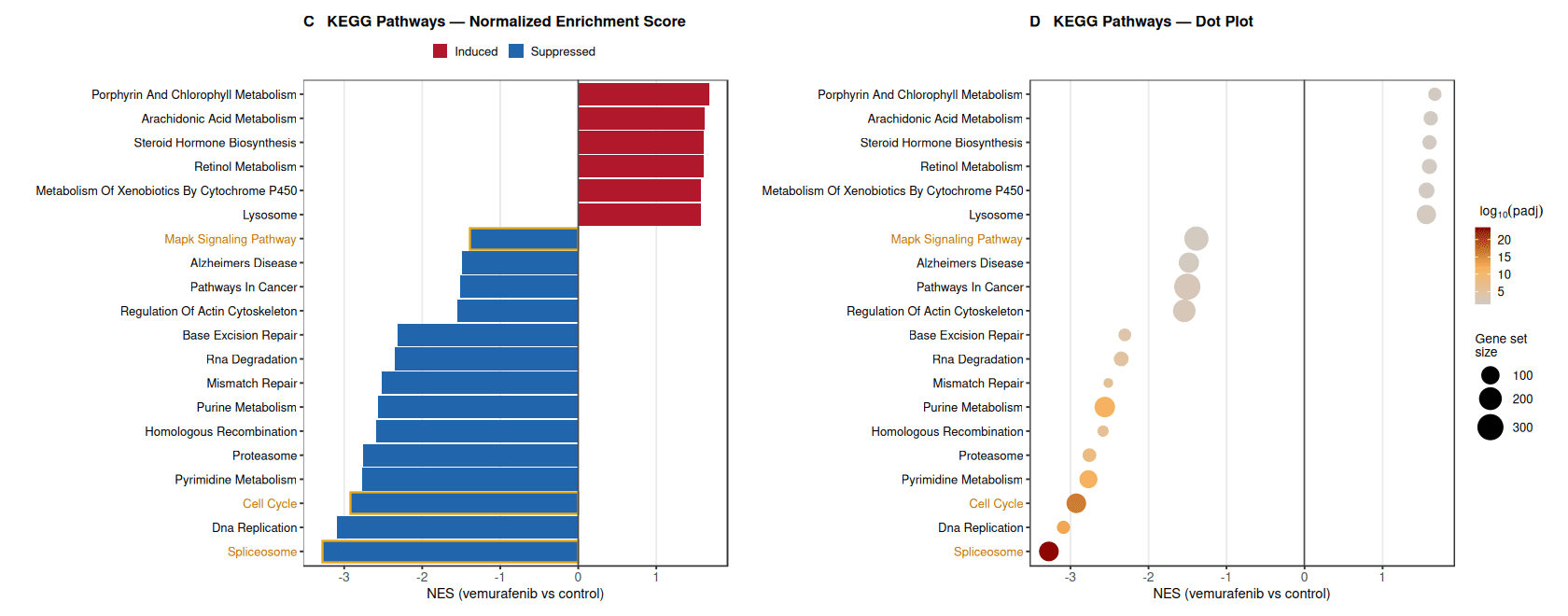
与这些观察结果相一致,使用KEGG路径数据库的路径富集进一步显示核心增殖和生物合成过程的强烈抑制。与细胞周期、DNA复制、剪接体和嘧啶代谢相关的基因集显示强烈的负规范化富集分数,暗示DNA合成和细胞周期进程受到抑制。
相反,几个代谢路径,包括维甲酸代谢、类固醇激素生物合成和细胞色素P450介导的外源代谢在治疗诱导的基因中相对富集。
总体来看,这些结果表明vemurafenib触发了一个协调的转录程序,特征是细胞周期和MYC驱动的增殖的抑制,以及压力和炎症信号路径的激活。此全球转录重编排与点一致,即ERK信号抑制导致增殖输出减少,同时促进补偿性信号程序以促使适应性抵抗。
干实验室确认的论文
•FGFR1和FGF2在HTH104和sw1736(甲状腺,SHP2阴性)中的富集:定量确认(tissue_diff = -1.93 和 -1.92)。
•ERBB3/AREG/EREG在WIDR(结直肠,SHP2阳性)中的富集:确认(tissue_diff = +1.82, +1.77, +1.92),支持了论文中描述的EGFR轴依赖性。
•FGFR同工型切换——甲状腺中FGFR1占优 vs. 结直肠WIDR中的FGFR3/4——是干实验室分析中新发现的定量结果,扩展了论文的FGFR1观察。
•vemurafenib时DUSP6和FOSL1的抑制:确认(排名列表中最被抑制的基因,t = -7.88 和 -8.26),为目标ERK抑制提供内部正控制。
干实验室的假设建议
新发现 | 生物学意义 |
|---|---|
EMT是最强的组织分裂分辨器 (NES = -3.86, padj = 6.5e-38 在甲状腺ATC中) | 未在论文中报告;与已确立的ATC生物学一致;解释了主要PC1方差的主导性 |
vemurafenib强烈抑制E2F Targets, MYC Targets V1/V2, G2M检查点 (NES -3.9 至 -4.3) | 论文未对RNA-seq数据进行路径级别分析;这是BRAF抑制的下游转录后果的完整描述 |
vemurafenib诱导干扰素Alpha/Gamma响应 (NES +2.29, +2.00) | MAPK抑制的免疫调节效应,未在原始论文中讨论;与临床试验数据(VEMUPLINT)相连 |
vemurafenib时剪接体的抑制 (NES = -3.28) | 可能因MYC抑制而次生;在原始论文中未报导;需要独立验证 |
vemurafenib时sw1736中的SOS1转录上调 | 建议在RAS-GEF级别上在甲状腺V600E细胞中发生转录补偿 |
V600E系中的NRG2诱导 | 潜在的ERBB3-PI3K绕过轴;在论文中未描述 |
V600E系中的FGFR2适度诱导 | 额外的FGFR同源复补偿 |
KEGG细胞周期富集正确分配给甲状腺ATC (NES = -2.74),不是WIDR | 解决了潜在的审稿人错误归因;考虑到ATC的极端有丝分裂指数,生物学上是一致的 |