1.2 干实验室资源概览
开始使用

Drylab 内置了无数资源,让您的分析更锐利、更快速、科学更加准确。从数据库和加速工具到完整的流程、预装的包和基于技能的工作流——每一层帮助 AI 理解您的意图,并提供与现实世界最佳实践一致的结果。
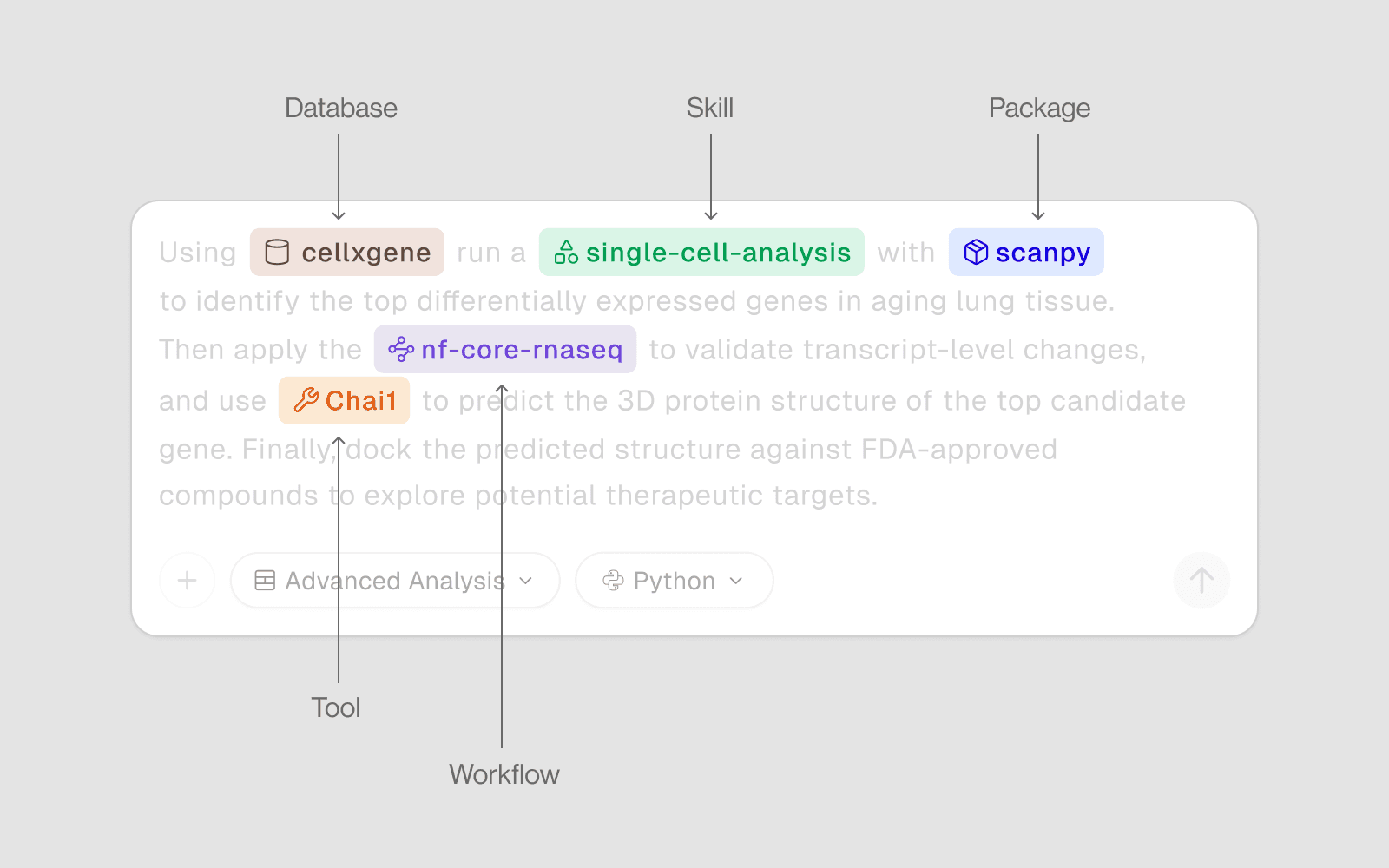
数据库 | 查询生物数据(不需要计算) |
加速工具 | 运行 GPU/HPC 任务(蛋白质折叠、对接) |
流程 | 端到端 Nextflow 工作流(RNA-seq,变异检测) |
流程 | Python/R 库(预装,即可使用) |
技能 | 分步分析指南(最佳实践工作流) |
概览
通过在聊天中使用 @ 直接调用这些资源,您可以指导 AI 使用正确的方法或数据集,使您的输出更加精确、可重复且完全符合您的期望。
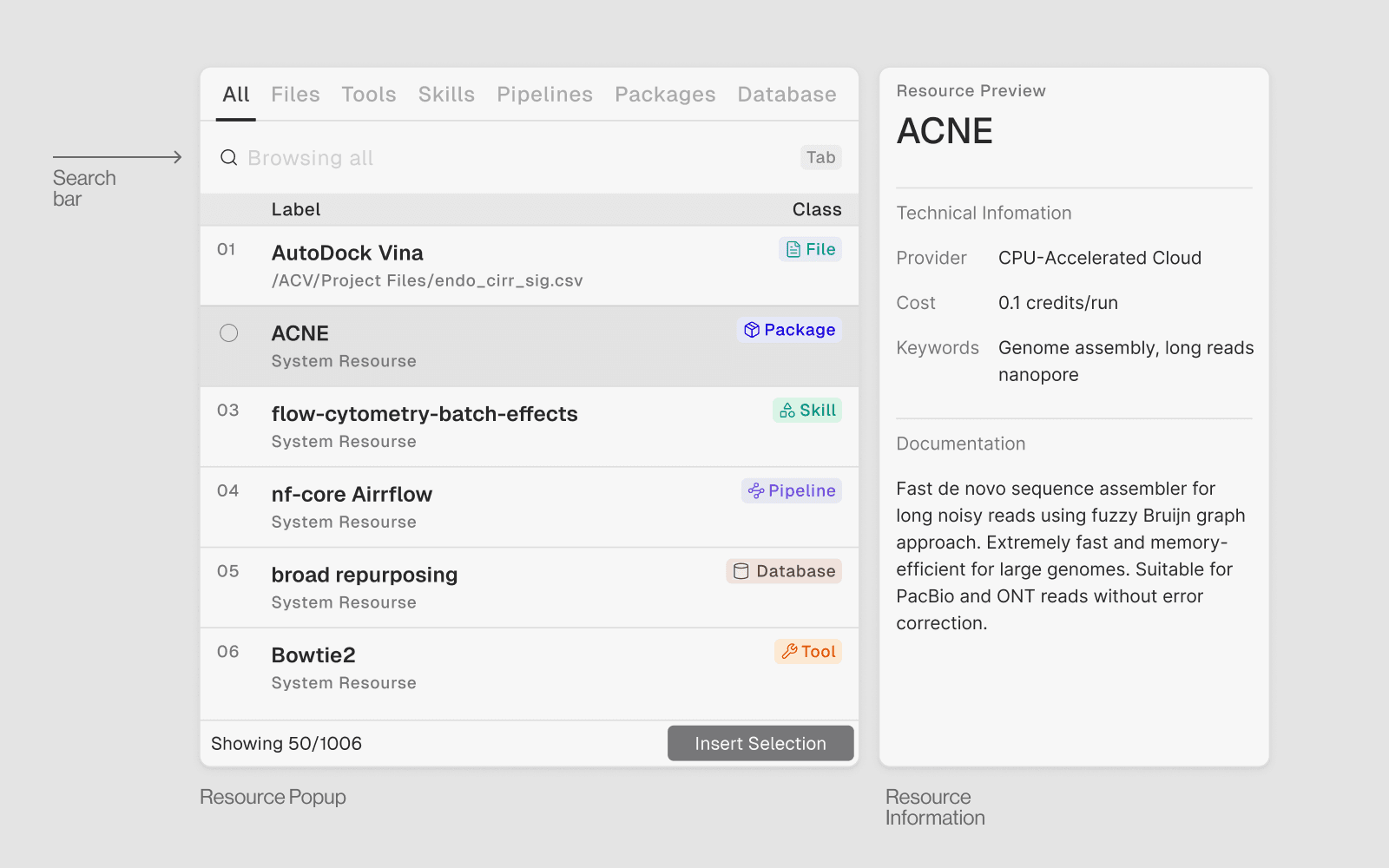
1. 数据库
它们是什么: 连接到 80 多个精选的生物数据库。用简单的英语查询它们——无需 API 密钥,无需手动 HTTP 请求。
何时使用: 当您需要参考数据时——基因信息、蛋白质结构、药物靶点、变异、通路、表达数据。
类别 | 关键数据库 |
|---|---|
蛋白质 | UniProt,PDB,AlphaFold |
基因组 | Ensembl,gnomAD,ClinVar |
通路 | KEGG,Reactome |
药物 | DrugBank,PubChem,ChEMBL |
癌症 | cBioPortal,OncoKB |
表达 | GTEx,GEO |
单细胞 | CellxGene |
2. 加速工具
它们是什么:
50 多种 GPU/HPC 提供的计算工具,适用于繁重的科学任务——蛋白质折叠、分子对接、基因组比对和变异检测。
何时使用:
当计算任务对标准 CPU 笔记本来说过于密集时——结构预测、大规模对接或长读长组装。
类别 | 工具 |
|---|---|
蛋白质折叠 | chai1, boltz2, ESMFold |
分子对接 | AutoDock Vina, DiffDock |
比对 | STAR, Minimap2, BWA |
组装 | Flye, SPAdes |
变异检测 | Clair3, Strelka2 |
示例:使用 Chai-1 预测此序列的 3D 结构:MKTAYIAKQR...
3. 流程
它们是什么:
84 个生产就绪的 Nextflow (nf-core) 工作流,用于完整的上游生物信息学处理——从原始 FASTQ 到分析准备输出。
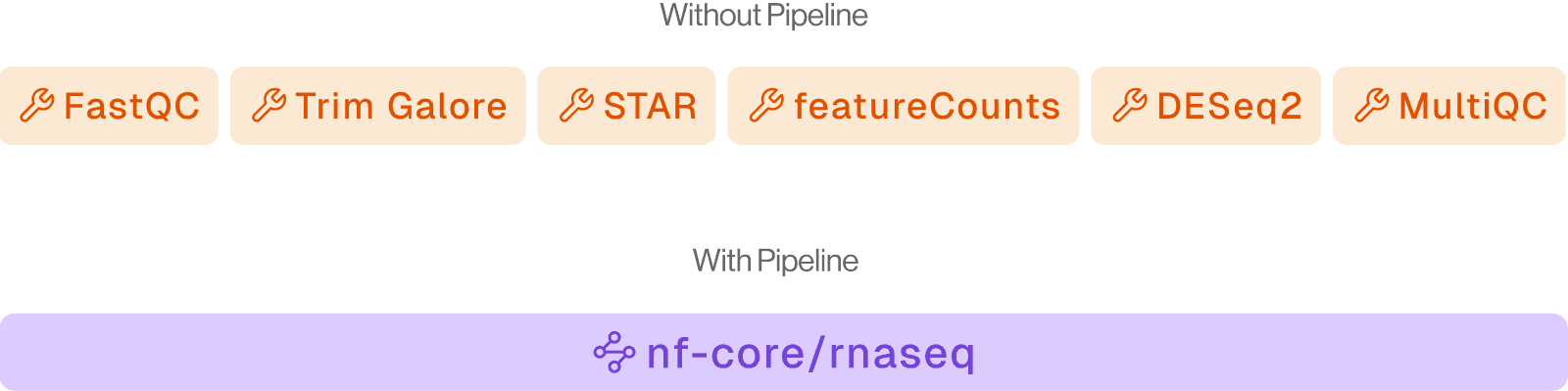
何时使用:
当您拥有原始测序数据并需要大规模的标准化、可重复处理时。
类别 | 流程 |
|---|---|
批量 RNA-seq | nf-core-rnaseq |
单细胞 | nf-core-scrnaseq |
变异检测 | nf-core-sarek |
扩增子 / 16S | nf-core-ampliseq |
ChIP-seq / ATAC-seq | nf-core-chipseq, nf-core-atacseq |
宏基因组学 | nf-core-mag, nf-core-taxprofiler |
示例:在 drylab://My Project/data/samplesheet.csv 上运行 nf-core-rnaseq 并使用基因组 GRCh38。
4. 包
它们是什么:
预装的 Python 和 R 库,覆盖完整的科学计算堆栈——无需设置即可导入。
何时使用:
对于笔记本中的所有交互分析——数据操作、统计、机器学习、可视化。
R 包(通过 Rscript 或 R 单元格):
Seurat | 单细胞 |
DESeq2 | 差异表达 |
ggplot2 | 可视化 |
edgeR | RNA-seq |
limma | 线性模型 |
示例:使用 Scanpy 对我的 h5ad 文件运行 PCA 和 Leiden 聚类,分辨率为 0.5。
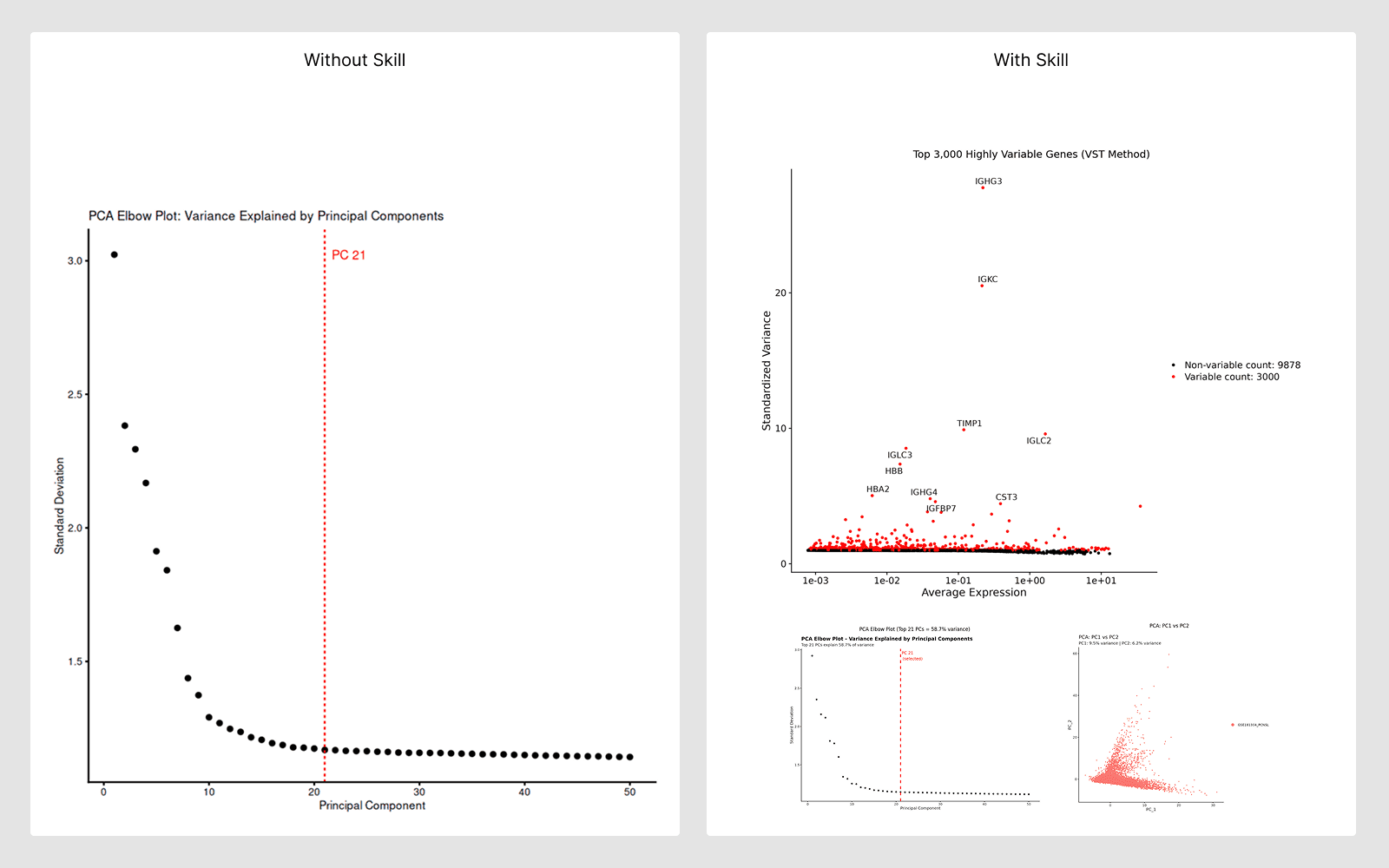
5. 技能
它们是什么:
精选的分步分析指南,编码特定工作流的最佳实践。AI 自动读取这些指南并遵循验证的协议。
何时使用:
在进行成熟的分析类型时——AI 遵循技能指南以确保科学上正确、可重复的结果。
领域 | 示例技能 |
|---|---|
单细胞 | QC,聚类,注释,DE,轨迹 |
空间 | 空间变量基因,去卷积 |
批量 RNA-seq | 标准化,DE,通路富集 |
蛋白质组学 | 结构预测,对接 |
系统发育 | 树重建,比对 |
AI 如何使用技能:
自动发现任务相关技能
读取协议,选择正确的方法和参数
根据技能的质量检查点验证结果
示例:根据最佳实践在我的 h5ad 文件上运行单细胞 QC 和聚类。
如何有效使用所有资源
让 AI 帮您发现资源
“我有 scRNA-seq 数据。应该使用哪些工具和数据库进行细胞类型注释和差异表达?”
将资源链接在一起
查询 CellxGene 获取人类肺参考图谱 → 数据库
使用 STAR 比对 FASTQ 文件 → 加速工具
运行 nf-core-scrnaseq → 流程
使用 Scanpy 和最佳实践聚类和注释 → 包 + 技能
查询 DrugBank 获取靶向识别标记的药物 → 数据库